☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
3_termodinámica_soluciones
Termodinámica de soluciones
Diapositiva 1
Diapositiva 1
Plata (Ag).
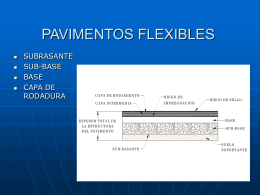
PAVIMENTOS FLEXIBLES
Cinética
Estrategia de la Copia de seguridad
Diapositiva 1
Diapositiva 1
RAZONAMIENTO CON INCERTIDUMBRE
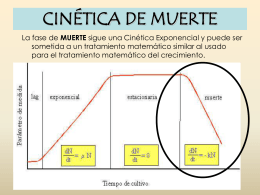
Cinética de Muerte
CINÉTICA QUÍMICA
El héroe, según la tragedia
El Hierro
Logaritmo y exponencial
Presentación de PowerPoint
Fundamentos de Hidrogeoquímica - Centro de Geociencias ::.. UNAM
Matematicas
Clase 129:Logarítmos Decimales - CubaEduca
Diapositiva 1
Diapositiva 1
Capacidad de un canal