☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
Salud
Click here to get the file
Aplasia Medula Anemia aplásica
Diapositiva 1
UNIVERSIDAD CATOLICA DE MANIZALES FACULTAD DE …
Anemia Aplástica Severa…
copia - admon.salud.campoalto
Caso 2. Varón de 15 años con astenia
Anemia por deficiencia de hierro
Anemia por déficit de hierro
Clasificacion de anemias, ferropenica, aplasica
PRESENTACION INUSUAL DE DEFICIT DE VITAMINA
Anemia - THER 2020
Presentación de PowerPoint
interpretación analítica del embarazo en atención primaria
Hemólisis adquirida - Departamento de Medicina Interna.
Interpretación-clínica-de-la-biometría
Hueso
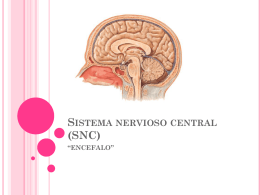
Sistema nervioso central
HIERRO
PROTEÍNAS: ÁNGELES O DEMONIOS
Curación ósea - Influencia del tratamiento
Leucemia - THER 2020
SISTEMA NERVIOSO