☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
Elaboración de un protocolo de ensayo clínico en
Slide 1
ETNOGRAFÍA DEL DIARIO DE CAMPO
ESPECIFICACIONES DE CALIDAD EN EL LABORATORIO …
gestion de control y ensayos del equipamiento aislante (003).
Document
El proceso de leer, responder y escribir
Diapositiva 1 - Bolsa de Valores de Lima
tipos de artículos - Portal de revistas académicas de la Universidad
Diapositiva 1
Diapositiva 1 - Sierra Exportadora
aspectos éticos, políticos, legales y morales al investigar
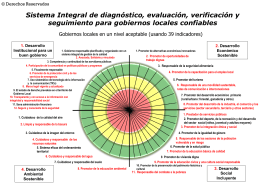
ISO 18091-2014-Cuadrantes
¿Cómo Aprenden nuestros hijos?
Metrología de fuerza aplicada a laboratorios de ensayo
ENSAYO CLINICO y AP
Leamos la Ciencia para Todos
PROPIEDADES MECÁNICAS
DISEÑOS DE INVESTIGACIÓN
Descargar Material - Consejo Profesional de Ciencias Económicas
Document
Tecnología apropiada (resultados)
auditoría 2010 material de apoyo que no reemplaza la lectura de la