☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
Salud
presentation
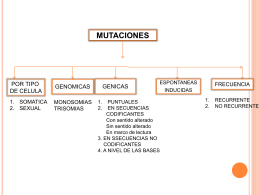
TIPOS DE MUTACIONES
mutaciones
Transformaciones societales de las últimas décadas
Biología Molecular
Traduccion-2008_Bioq_100
REDES NEURONALES ARTIFICIALES
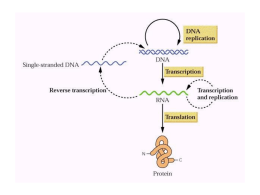
Replicación
Diferencial Geneticas Parkinsonismos
ppt 1Origen de la vida y evolucion
Alfa thalassaemia
4.1 Tipos y funciones de RNAs
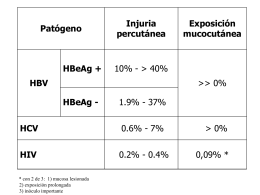
HIV
Determinación del factor de carga de pérdidas de energía en redes