☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
Salud
Diferencial Geneticas Parkinsonismos
Diapositiva 1
Atrofia Multisistemica
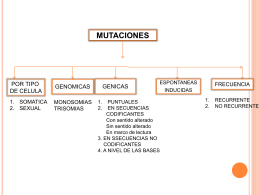
mutaciones
Descargar documento de la ponencia
Me llamo Chris. Yo fue
Sdsadsad sadsadasdasd
presentation
Transformaciones societales de las últimas décadas
MOVIMIENTOS ANORMALES
Técnicas de Calidad en el Software
UNIDAD II Métricas y Procesos PSP Personal
Alfa thalassaemia