☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
HERENCIA NO TRADICIONAL
Las cegueras del conocimiento: El error y la ilusión - DHPC
Inteligencia Artificial
Sesión 7ª
BASES MOLECULAR Y BIOQUÍMICA DE LAS ENFERMEDADES
Diapositiva 1
Heuristicas para la optimizacion
Haga clic aquí para descargar la documentación del
PROYECTO GENOMA HUMANO
Cultivos Transgénicos: ¿Frankenfood vs. Robin Food?
Enfermedad neurodegenerativa por almacenamiento de
IMPRONTA GENOMICA (“IMPRINTING”)
SEMINARIO
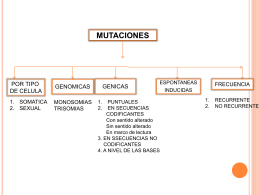
mutaciones