☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
SINDROME DE ROTHMUND THOMSON TIPO I: REPORTE …
catarata folleto
Instituto Regional de Oftalmología
Diapositiva 1
PSICOLOGIA DEL COLOR
Síndrome de Down (trisonomía 21)
CATARATAS PREGUNTAS Y RESPUESTAS
File - Zuleika Ponce
File
Sindrome de Dawn vb
Descarga
SINDROME DE TURNER El Síndrome de Turner definido como el
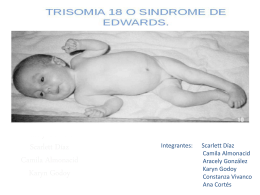
Síndrome de Edwards
SÍNDROME DE TURNER
PROCEDIMIENTO: Paso 1: Imprimir 2 copias del caso que elegiste
ALTERACIONES HUMANAS - elmatraz
Diapositiva 1
Publicado en BIREME (OPS) y en Archivos de Oftalmologia
Slide 1
Catarata Congénita - Pontificia Universidad Javeriana