☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
Termo 8
No Slide Title
Diseño de transparencias
APLICACIÓN DE LA DERIVADA A OTRAS ÁREAS
TERMO2005 CAP9 – Primera Ley Sistemas Cerrados Mayo 2005
TERMO2005 CAP10 – Primera Ley Sistemas
7 Movimientos en caída libre y resistencia al viento
Descarga
TERMO2005 CAP8 – Trabajo y Calor Abril 2005
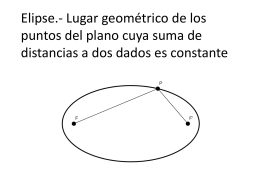
Elipse.- Lugar geométrico de los puntos del plano cuya suma de
Decent work for domestic workers
Canales de marketing y ventas al detalle
Risk-adapted therapy in APL (Stockholm, August