☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
Práctica 1: Alineamientos
Diapositiva 1 - Universidad de Antofagasta
METODOS DE DISTANCIA
Diapositiva 1 - Universidad de Antofagasta
Escalogramas multidimensionales
Bayesianos - Instituto de Biología
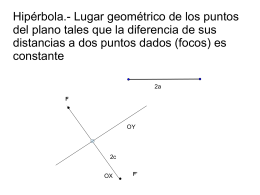
Hipérbola.- Lugar geométrico de los puntos del plano tales que la
Diapositiva 1
Presentación PowerPoint de los métodos de reconstrucción
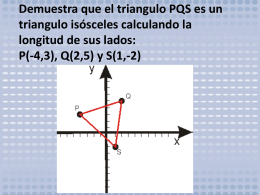
Demuestra que el triangulo PQS es un triangulo isósceles
Estrategia - asesorlinguistico02
Ubicación de tuberías en una instalación industrial
Construcción de árboles filogenéticos
Estimación de máxima verosimilitud
¿Qué es el método de estimación de máxima verosimilitud y cómo
PARTE 1 (Archivo ppt)
Alcances probatorios negativa del demandado a someterse a las
Trabajo: Proyecto de medición
examen diagnóstico - Instituto de Investigaciones Filosóficas
Diapositiva 1
MEDIDAS CAUTELARES