☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
Diapositiva 1
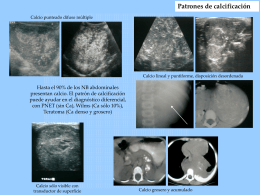
4. Patrones de calcificación
Prueba de Esfuerzo File
angina
la enfermera especialista en insuficiencia cardiaca
Nomenclatura y notación química de las sales ternarias
(I). Etiología, fisiopatología,clasificación y diagnóstico. (Pilar Sampériz)
3) La perspectiva clínica en la estratificación pronóstica. Los
insuficiencia cardiaca
Alimentos cardiosaludables
Programa de Falla Cardiaca Clínica Universitaria
Taller 2 Centrado en el tratamiento intra (descompensación IC)