☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
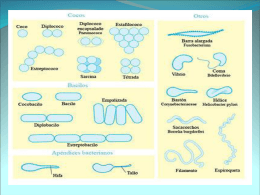
Diapositiva 1 - LGVH
TWITTER Redireccionamiento en el sentido de las frases:
TECNICAS PARA ANALISIS DE ACIDOS NUCLEICOS
Lab. #1: Base Molecular de la Herencia
Document
Lab. #1: Base Molecular de la Herencia
Diapositiva 1
Components of a PCR and RAPD Reactions
FACTORES QUE DETERMINAN LA SENSIBILIDAD Y LA …