☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
Ciencia
Diapositiva 1
Principales errores cometidos por emprendedores
SIMULACIÓN CON EL SOFTWARE ARENA
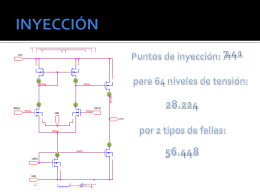
Inyección de fallas en estructuras analógicas CMOS
Presentado en informatica - informaticaeducativaunah-vs
Simulación Clínica como Metodología de la Enseñanza Modelo
Slide 1
Simul8 - saavedracomputacion1 / FrontPage
SINDROME DE VENA CAVA SUPERIOR