☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
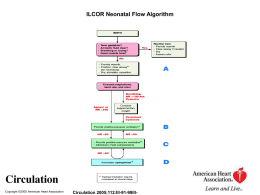
Diapositiva 1
Cribado Neonatal Comunidad Madrid
Descargar Programa en formato PDF
Cribado neonatal Murcia (2012)
La Mortalidad Infantil en Costa Rica
El programa de salud infantil ¿qué, quién, cuántas veces?
Actividades preventivas en los ancianos
impacto de una campaña educacional sobre la adherencia al
instrumentos de cribado de los trastornos del desarrollo psicomotor
Estrategia regional para la cobertura universal de salud. Reunión
Resumen mesa PAPPS
INFORMAZIO ESKE: Nerea Antía Vinós andreak, Euzko Abertzaleak
Diapositiva 1
Diapositiva 1 - Home - Pan American Health
Document
ENFERMEDADES ORGANOESPECIFICAS AUTOINMUNES
Cribado cromosomopatías en Castilla
ACTIVIDADES PREVENTIVAS EN ATENCION PRIMARIA
Mortalidad I
papps- (2007)
Protocolo de Cribado Neonatal de Enfermedades Endocrino
Recomendaciones Preventivas 2005
aciduria glutárica tipo-i