☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
Salud
Diapositiva 1
Tríptico de la Gangliosidosis GM1
Presentación de PowerPoint
Espejos de corriente - Electrical and Computer
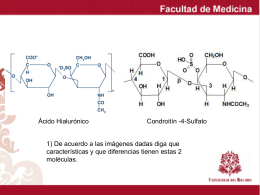
carbohidratos que forman parte de la matriz
Descargar PDF - Revista Mexicana de Neurociencia
Efecto Vodka - Curso de Sistema Financiero Internacional
Diapositiva 1 - DISASTER info DESASTRES
EFECTO VODKA - Curso de Sistema Financiero Internacional
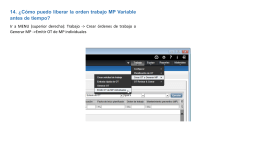
Cómo puedo liberar la orden trabajo MP Variable antes de tiempo?
Diapositiva 1
utilizacion del monosialogangliosido gm1 o del n-dicloroacetil
El niño como sujeto de derecho
Dispositivos para Filtrar el Agua