☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
Síndrome de Down Neurologia
(CASO CLÍNICO): Síndromes Glomerulares
Síndrome de Goodpasture
Palestra I
contrato de adesão à fase de teste do serviço giro beta
CASO CLÍNICO SÍNDROME NEFRÓTICA
Caso Clinico: Síndrome de Guillain-Barré
LINFADENOMEGALIAS
Manual da fase de teste do Giro Beta
Interpretação do hemograma em pediatria
Obesidade e Dislipidemia - Dr. Rodinei 27/05/2008
Síndrome Fetal Alcoólica - José Salomão Schwartzman
COMO CALCULAR O COEFICIENTE BETA DE UMA EMPRESA DE
INDICADORES DE CAPITAL DE GIRO E BETA: UM ESTUDO NO
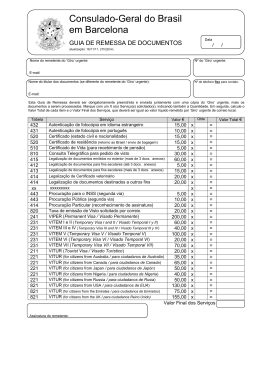
Consulado-Geral do Brasil em Barcelona
DX53W Escopo de Trabalho
xxi congresso brasileiro de adolescência
Síndrome de Rett - José Salomão Schwartzman
Síndrome de Rett - José Salomão Schwartzman