
Contenido
Doradobet en Honduras: guía para apuestas y casino online
Doradobet es una marca que muchos usuarios hondureños buscan cuando quieren conocer una plataforma de apuestas deportivas y casino online antes de registrarse, depositar o reclamar un bono. La experiencia puede parecer sencilla desde fuera, pero en la práctica conviene revisar varios puntos importantes: cómo funciona la cuenta, qué se puede hacer en la sección deportiva, qué juegos de casino están disponibles, cómo se activan los bonos, qué métodos de pago aparecen en la caja y qué medidas de seguridad debe tomar el jugador.

image placeholder 1
Esta guía está pensada para explicar qué es doradobet Honduras y cómo funciona para apuestas y casino online, cómo leer el bono de bienvenida de Doradobet en Honduras: requisitos, rollover y límites, qué revisar sobre métodos de pago en Doradobet Honduras: Tigo Money, BAC, tarjetas y retiros, y cómo usar Doradobet app y versión móvil en Honduras: funciones, seguridad y soporte. El objetivo no es prometer ganancias ni presentar la plataforma como una solución financiera, sino ayudar al usuario a tomar decisiones con más información.
Antes de usar dinero real, es recomendable mirar Doradobet como una cuenta digital completa. No se trata solo de elegir un partido o abrir una tragamonedas. También hay que proteger el acceso, revisar el presupuesto, leer condiciones, confirmar límites y entender que toda apuesta o juego de casino implica riesgo. Un usuario que revisa primero y juega después suele evitar muchos errores comunes.
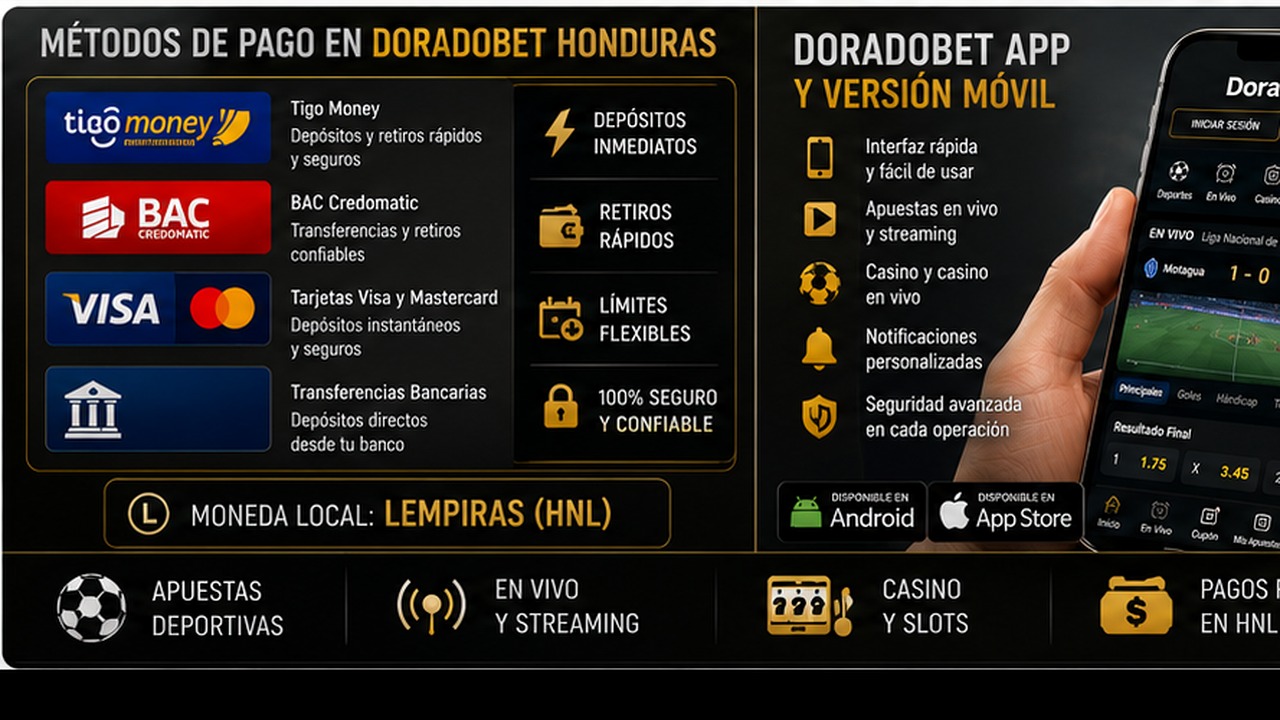
image placeholder 2
Qué es Doradobet Honduras y cómo funciona para apuestas y casino online
Qué es Doradobet Honduras y cómo funciona para apuestas y casino online es la primera duda de muchos usuarios nuevos. En términos generales, Doradobet puede entenderse como una plataforma donde el jugador crea una cuenta, accede a apuestas deportivas, revisa juegos de casino online, consulta promociones y gestiona depósitos o retiros desde su perfil. La disponibilidad exacta de secciones, métodos y funciones puede depender del estado de la cuenta, la región y la configuración actual de la plataforma.
El recorrido habitual empieza con el registro. El usuario introduce datos personales, crea una contraseña y accede a su perfil. Desde ahí puede revisar deportes, mercados, casino, bonos, caja y soporte. Lo importante es que cada sección tiene reglas propias. Una apuesta deportiva no se evalúa igual que una ronda de tragamonedas. Un bono no funciona igual que saldo libre. Un retiro puede requerir más controles que un depósito.
Cuenta y perfil del usuario
La cuenta personal es el centro de la experiencia en Doradobet. Desde ahí se administra el saldo, se revisan promociones, se consultan pagos y se contacta con soporte. Por eso conviene registrarse con datos reales y actuales. Si más adelante se solicita verificación, la información del perfil debe coincidir con los documentos del usuario.
- usar un correo electrónico o teléfono activo
- crear una contraseña única y segura
- evitar datos incompletos o incorrectos durante el registro
- mantener el acceso al correo y al número registrado
- revisar el perfil antes de hacer el primer depósito
Estos pasos parecen simples, pero ayudan a prevenir problemas posteriores. Una cuenta con datos claros facilita la recuperación de acceso, el contacto con soporte y las posibles verificaciones antes de un retiro.
Apuestas deportivas en Doradobet
La sección deportiva suele organizarse por deportes, competiciones, eventos, mercados y cuotas. El usuario puede elegir un partido, revisar las opciones disponibles y añadir una selección al boleto. Antes de confirmar, debe comprobar el mercado, la cuota y el monto apostado. Si la cuota cambia antes de aceptar, es mejor revisar el boleto nuevamente.
- elegir deporte y competición
- abrir el evento disponible
- leer el mercado antes de seleccionar
- confirmar la cuota en el boleto
- apostar solo dentro del presupuesto definido
En apuestas deportivas, entender el mercado es tan importante como elegir el evento. Un hándicap, un total de goles o una apuesta en vivo tienen reglas distintas. Apostar sin comprender la liquidación puede causar confusión cuando el evento termina.
Casino online en Doradobet
El casino online puede incluir tragamonedas, ruleta, blackjack, juegos de mesa y opciones con crupier en vivo, según lo que aparezca disponible en la cuenta. Cada juego tiene una mecánica propia. Las tragamonedas suelen tener rondas rápidas, mientras que los juegos en vivo exigen más atención al ritmo de la mesa y a la conexión.
- leer reglas del juego antes de apostar
- revisar apuesta mínima y máxima
- probar demo si está disponible
- comprobar si el juego cuenta para un bono activo
- evitar subir la apuesta por emoción del momento
El casino online puede ser entretenido, pero siempre implica riesgo. La mejor práctica es separar el presupuesto de apuestas deportivas del presupuesto de casino para no mezclar decisiones.
Bono de bienvenida de Doradobet en Honduras: requisitos rollover y límites
El bono de bienvenida de Doradobet en Honduras: requisitos, rollover y límites puede ser una de las primeras promociones que mira un usuario nuevo. Sin embargo, un bono no debe aceptarse solo por el monto anunciado. Toda promoción tiene reglas. Puede requerir depósito, activación manual, código promocional, cuota mínima, juegos elegibles, fecha de vencimiento o rollover.
El punto más importante es entender que el bono no siempre equivale a saldo libre. Puede estar separado del saldo real y exigir condiciones antes de ser retirado. Por eso el usuario debe leer los términos completos antes de confirmar un depósito o iniciar una apuesta vinculada a la promoción.
Qué revisar antes de activar un bono
Antes de aceptar el bono de bienvenida, conviene revisar si la promoción está disponible para la cuenta hondureña, si requiere un depósito mínimo, si aplica solo a casino o apuestas deportivas, y si hay límites de retiro o apuesta máxima. Cada detalle puede cambiar la utilidad real del bono.
- verificar si el bono está disponible para nuevos usuarios
- leer si requiere depósito o código promocional
- comprobar la fecha de vencimiento
- revisar si aplica a deportes, casino o ambos
- mirar la apuesta máxima permitida con saldo de bono
- confirmar si existen límites de retiro o ganancia máxima
Si una condición no está clara, es mejor consultar soporte antes de activar la promoción. Depositar primero y preguntar después puede generar una experiencia incómoda si el método usado no califica para el bono.
Cómo entender el rollover
El rollover indica el volumen de juego o apuesta que puede exigirse antes de retirar fondos vinculados a un bono. La fórmula básica para entenderlo es sencilla:
Si una promoción usa como base 500 HNL y exige rollover x10, el volumen teórico sería de 5.000 HNL. Si la base incluye depósito más bono, el volumen puede ser mayor. Este ejemplo solo explica la lógica. La condición real debe leerse en la promoción activa de Doradobet.
Límites y errores comunes
Los límites pueden incluir monto máximo de apuesta, retiro máximo, tiempo de uso, mercados válidos o juegos elegibles. Muchos usuarios cometen errores porque miran solo el título del bono y no abren las reglas completas.
- usar el bono en juegos no elegibles
- apostar con una cuota menor a la exigida
- olvidar la fecha de vencimiento
- superar la apuesta máxima permitida
- pensar que el bono se puede retirar de inmediato
El bono puede ser útil si el usuario entiende sus condiciones. Si obliga a apostar de una forma incómoda o demasiado arriesgada, puede ser mejor no activarlo.
Métodos de pago en Doradobet Honduras: Tigo Money BAC tarjetas y retiros
Métodos de pago en Doradobet Honduras: Tigo Money, BAC, tarjetas y retiros es un tema clave para jugadores que quieren usar saldo real. Antes de depositar, el usuario debe abrir la caja de su perfil y revisar qué opciones aparecen disponibles. Es importante no asumir que un método estará activo para todos los usuarios o en todas las condiciones. La disponibilidad puede depender de la cuenta, la región, el estado de verificación y las reglas vigentes.
Tigo Money, BAC, tarjetas y otros métodos pueden interesar a usuarios hondureños por familiaridad local, pero cada opción debe comprobarse directamente en la caja. También hay que revisar mínimos, máximos, posibles comisiones, tiempo de procesamiento y si el método sirve para depósitos, retiros o ambos.
Depósitos con métodos locales
El depósito es el paso que permite jugar con dinero real. Antes de confirmar una operación, conviene verificar que el método elegido sea compatible con la cuenta y que el monto se encuentre dentro de los límites visibles. Si se busca activar un bono, también hay que comprobar si ese método participa en la promoción.
- abrir la caja desde el perfil
- comprobar si Tigo Money aparece disponible
- revisar si BAC u opciones bancarias están habilitadas
- confirmar si las tarjetas son aceptadas para la cuenta
- leer límites mínimos y máximos
- verificar si el método activa bonos
Un depósito bien revisado evita errores simples. El usuario debe confirmar datos, monto y condiciones antes de enviar el pago. Si la operación tiene conversión o comisión, debe entenderla antes de aceptar.
Retiros y verificación
Los retiros pueden requerir más pasos que los depósitos. La plataforma puede revisar identidad, método usado, estado del bono y coherencia de datos. Por eso es importante que el perfil esté completo y que el método de pago pertenezca al mismo usuario.
- usar métodos de pago propios
- comprobar si el método permite retiros
- revisar límites de retiro visibles
- confirmar si hay KYC pendiente
- verificar si el rollover del bono está completo
- contactar soporte si el retiro queda pendiente demasiado tiempo
Un retiro puede tardar por revisión interna, procesamiento del método, verificación de documentos o condiciones promocionales. No siempre se trata de un problema grave. Lo mejor es revisar el estado de la solicitud antes de repetir operaciones.
Tiempo de procesamiento y expectativas realistas
Los tiempos pueden variar según método, perfil y verificaciones. Un usuario con cuenta recién creada puede enfrentar más controles que uno con datos ya confirmados. También puede haber diferencias entre depósito y retiro. Por eso la caja y los mensajes de soporte deben considerarse la fuente principal de información actual.
La expectativa más saludable es entender que rapidez y disponibilidad no son iguales para todos los métodos. Antes de elegir una opción, el usuario debe preguntarse si puede usarla también para retirar y qué requisitos se aplican.
Doradobet app y versión móvil en Honduras: funciones seguridad y soporte
Doradobet app y versión móvil en Honduras: funciones, seguridad y soporte es una búsqueda importante porque muchos usuarios prefieren apostar o jugar desde el teléfono. La versión móvil permite entrar desde el navegador sin instalar nada. La app, si está disponible para el usuario, puede ofrecer acceso más directo y una interfaz adaptada a pantalla pequeña. La elección depende del dispositivo, del uso frecuente y de la confianza en la fuente de instalación.
El móvil facilita la navegación, pero también puede aumentar la velocidad de las decisiones. Por eso conviene mantener los mismos controles que en computadora: revisar el boleto, leer bonos, comprobar pagos y cerrar sesión si se usa un dispositivo ajeno.
Funciones de la versión móvil
La versión móvil suele permitir acceso a cuenta, deportes, casino, promociones, caja y soporte desde el navegador. Es útil para usuarios que no quieren instalar aplicaciones adicionales o que entran de manera ocasional. La principal precaución es revisar siempre el enlace antes de iniciar sesión.
- no requiere instalación
- funciona desde navegador actualizado
- permite revisar cuenta y saldo
- facilita acceso a deportes y casino
- exige verificar dominio antes de ingresar datos
La versión móvil puede ser suficiente para muchos usuarios. Si el sitio carga bien y el dispositivo es seguro, no siempre hace falta instalar una app.
App móvil y compatibilidad
Si Doradobet app está disponible para Android o iOS, el usuario debe instalarla solo desde una fuente confiable. También debe revisar compatibilidad, espacio disponible, permisos solicitados y actualizaciones. Descargar archivos desde enlaces desconocidos puede poner en riesgo la cuenta.
- verificar la fuente de descarga
- comprobar compatibilidad con el sistema
- mantener el teléfono actualizado
- revisar permisos antes de instalar
- proteger el dispositivo con PIN o biometría
La app puede ser cómoda para usuarios frecuentes, pero la comodidad no debe reemplazar la seguridad. Si la fuente no es clara, es mejor usar la versión móvil oficial o consultar soporte.
Seguridad y soporte desde el móvil
El usuario debe proteger la cuenta con contraseña fuerte, dispositivo bloqueado y acceso controlado al correo o teléfono registrado. Si aparece actividad sospechosa, conviene cambiar la contraseña y contactar soporte. Para reportar problemas, el mensaje debe incluir datos claros sin compartir información sensible.
- no compartir códigos OTP
- cerrar sesión en dispositivos compartidos
- evitar redes Wi-Fi públicas para pagos
- contactar soporte desde canales oficiales
- no enviar contraseñas ni datos completos de tarjeta
El soporte puede ayudar con pagos, bonos, acceso, verificación o fallos de la app. Un mensaje claro y seguro facilita la solución.
Cuotas mercados y apuestas en vivo en Doradobet
En la sección deportiva de Doradobet, el usuario debe entender cuotas, mercados y apuestas en vivo. Las cuotas muestran el precio de un resultado. Los mercados indican qué se apuesta. En vivo, estos elementos cambian con rapidez porque el evento ya está en desarrollo.
La fórmula básica para cuotas decimales es:
Si una persona apuesta 100 HNL a cuota 2.00, el retorno posible sería 200 HNL si gana. Este ejemplo solo explica la mecánica. No significa que una selección sea segura.
- leer el mercado antes de seleccionar
- revisar si la cuota cambió antes de confirmar
- evitar apuestas en vivo por impulso
- no perseguir pérdidas durante el mismo partido
- mantener límites separados para apuestas en vivo
Las apuestas en vivo pueden ser entretenidas, pero también más emocionales. El usuario debe confirmar solo cuando entiende el mercado y acepta el riesgo.
Casino online en Doradobet
El casino online puede incluir tragamonedas, juegos de mesa y juegos con crupier en vivo según disponibilidad. Cada tipo de juego exige una lectura diferente. Las tragamonedas pueden tener ritmo rápido y funciones especiales. La ruleta y el blackjack en vivo requieren conocer reglas y límites de mesa.
- revisar reglas antes de jugar
- mirar apuesta mínima y máxima
- usar demo si está disponible
- evitar subir apuestas para recuperar pérdidas
- separar presupuesto de casino y apuestas deportivas
El casino debe tratarse como entretenimiento. Ningún juego garantiza resultados, y las promociones no eliminan el riesgo de pérdida.
Gestión del presupuesto en Doradobet
La gestión del presupuesto es esencial en Doradobet. Antes de apostar o jugar, el usuario debe decidir cuánto dinero puede destinar al entretenimiento sin afectar gastos importantes. Ese límite no debe cambiar por una racha positiva ni por una pérdida reciente.
Una fórmula simple ayuda a medir el riesgo:
Si el presupuesto de sesión es de 1.000 HNL y una apuesta es de 100 HNL, esa jugada representa 10 por ciento del presupuesto. Esta lectura ayuda a evitar montos demasiado altos para una sola decisión.
- definir presupuesto antes de iniciar sesión
- usar montos proporcionales al saldo de entretenimiento
- no aumentar apuestas después de perder
- hacer pausas si la decisión se vuelve emocional
- no usar dinero destinado a obligaciones personales
La gestión del presupuesto no garantiza ganancias. Su función es mantener el riesgo dentro de límites definidos por el propio usuario.
Juego responsable
Doradobet debe usarse como entretenimiento, no como forma de resolver problemas económicos. Las apuestas deportivas, el casino online, los bonos y el cashback pueden resultar atractivos, pero no eliminan el riesgo. El usuario debe jugar solo con dinero destinado al ocio y mantener límites de tiempo y presupuesto.
Si aparece ansiedad, necesidad de recuperar pérdidas o dificultad para detenerse, es recomendable hacer una pausa. Si la plataforma ofrece límites, periodos de descanso o autoexclusión, estas herramientas pueden ayudar. Las apuestas y el casino online son solo para personas adultas.
Doradobet: conclusión para Honduras
Doradobet puede interesar a usuarios hondureños que buscan apuestas deportivas y casino online, pero conviene entender cada parte antes de usar dinero real. Qué es Doradobet Honduras y cómo funciona para apuestas y casino online ayuda a ubicar la cuenta, los deportes, el casino y los pagos. Bono de bienvenida de Doradobet en Honduras: requisitos, rollover y límites recuerda que ninguna promoción debe activarse sin leer condiciones.
Métodos de pago en Doradobet Honduras: Tigo Money, BAC, tarjetas y retiros muestra la importancia de revisar la caja y no asumir disponibilidad general. Doradobet app y versión móvil en Honduras: funciones, seguridad y soporte explica que la comodidad del teléfono debe ir acompañada de protección de cuenta. El mejor enfoque es verificar, leer, apostar con límites y pedir soporte cuando una regla no esté clara.
FAQ
Qué es Doradobet
Doradobet es una plataforma vinculada a apuestas deportivas y casino online, donde el usuario puede crear una cuenta, revisar eventos, jugar, consultar promociones y gestionar pagos según las opciones disponibles.
Qué es Doradobet Honduras y cómo funciona para apuestas y casino online
Funciona mediante una cuenta personal desde la que el usuario accede a deportes, casino, bonos, caja y soporte. Cada sección tiene reglas propias que conviene leer antes de usar dinero real.
Bono de bienvenida de Doradobet en Honduras: requisitos rollover y límites qué revisar
Hay que revisar disponibilidad, depósito mínimo, rollover, fecha de vencimiento, juegos o mercados elegibles, apuesta máxima y posibles límites de retiro antes de activar el bono.
Métodos de pago en Doradobet Honduras: Tigo Money BAC tarjetas y retiros están siempre disponibles
No necesariamente. El usuario debe revisar la caja de su perfil para confirmar si Tigo Money, BAC, tarjetas u otros métodos están disponibles para depósitos, retiros o ambos.
Doradobet app y versión móvil en Honduras: funciones seguridad y soporte qué diferencia hay
La versión móvil funciona desde el navegador sin instalación. La app puede ofrecer acceso más directo si está disponible, pero debe instalarse solo desde una fuente confiable y con el dispositivo protegido.
Doradobet garantiza ganancias
No. Ninguna plataforma de apuestas o casino online garantiza ganancias. El usuario debe jugar solo como entretenimiento, con presupuesto definido y sin perseguir pérdidas.