☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
Document
SIMULACIÓN CON EL SOFTWARE ARENA
LOS GASES
Estados de la materia”
GUÍA LÍQUIDO
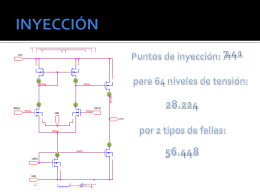
Inyección de fallas en estructuras analógicas CMOS
Presentado en informatica - informaticaeducativaunah-vs
SUELO
Diapositiva 1
Unidad I:La materia y sus Transformaciones
INTRODUCCIÓN A LA FÍSICA