☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
POLIPOS EN COLON Y RECTO
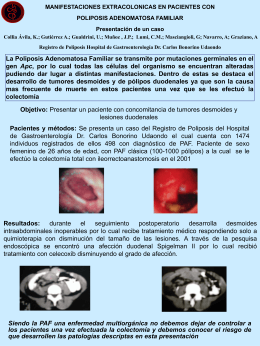
Manifestaciones Extracolónicas en Pacientes con Poliposis
No Slide Title
11. Cancer de colon sospecha (PPTminimizer)
Los Cnidarios
Clasificaciones en Endoscopía - Endoscopia UC
RUTAS DE LOS VIAJES DE COLON
Lesiones benignas de vulva vagina y cervix
Cáncer Gástrico: Lesiones Incipientes
Diapositiva 1
Diapositiva 1 - WordPress.com
Viajes de Colón
Archivo (33 segundos a 56 Kb/s)
Descarga - Educacion preescolar zona 33