☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
Comparación de secuencias
Homologous sequences - Molecular Modeling and Bioinformatics
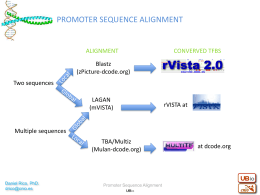
Promoter sequence alignment
Document
Document
Aucun titre de diapositive - Institut de Recherche en
No Slide Title
Toward mapping listening skills on the CEFR: An
Document
Document
Data Mining: Concepts and Techniques — Chapter 8 — 8.2
SER V. ESTAR
Escribe el nombre de una person famosa para cada adjetivo:
Positive and Negative Feedback Mechanisms in the
Molecular genetics of gene expression
Automatic Application-Specific Instruction
My history of women
SER vs. ESTAR
An Introduction to Machine Translation
Role of unequal-crossover in alpha
El Pretérito de los verbos ser y estar PPT
ser-vs-estar-doctor-and
Parte 2