☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
LAS ENFERMEDADES HEMORRAGICAS
19. la gran ramera - Iglesia Cristiana La Serena
presentacion-psicologia-del-color-5
PURPURA - Seccionseis’s Weblog
REINO PLANTAE - Colegio Nuestra Señora
la inocencia
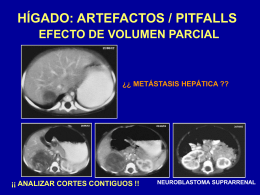
Figura 8. Artefactos / Pitfalls relacionados con el hígado.
alvaro abad
Economia y Empresa, Ralph Pochadt
Compare ambas estructuras. Coloque labelos. Indique grupos de
Enfermedades Raras