☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
Presentación de PowerPoint
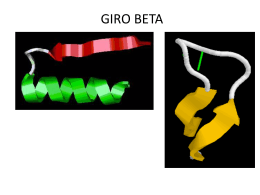
GIRO BETA - e
DIVERSIDAD α β γ
Beta - Gaceta Financiera
Lámina Beta y Alfa Hélices.
Placebo/second sub standard and resource poor settings
movilizacion cervical y dolor de hombro
Construcción Portal Web Institucional (Internet
Estadística básica
no validada - StudentConsult.es
Costo de capital (presentación Power Point)