☰
Explorar
Iniciar sesión
Crear una nueva cuenta
Pubblicare
×
Descargar
No category
Alfa thalassaemia - Blog de Química Biológica
Alfa thalassaemia
Diapositiva 1
comic ana, luz
Los Simpson - Mariajosecubillos9
Diapositiva 1
Diapositiva 1
Presentación de PowerPoint
la_casa_de_los_simpsons[1]
La carrera de los Simpson
Bart Simpson esta enfermo story
PROBLEMAS DE GENÉTICA
Document
Comando de VBA para PowerPoint
Beta talasemias - Blog de Química Biológica
Ma famille
Diapositiva 1
Rasgo de Alfa Talasemia - St. Jude Children`s Research Hospital
Rasgo de Alfa Talasemia
Libro 2
Enfermedades relacionadas con los aminoácidos
capítulo 3-2 - Web Médica Argentina

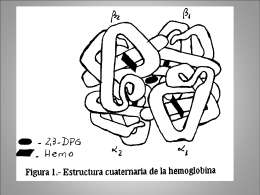




![la_casa_de_los_simpsons[1]](http://s1.slideshowes.com/store/data/000036434_1-2c3ae9b008dcbd3e96162010487a4a83-260x520.png)












